Pipelines¶
Pipelines are pre-configured Nextflow workflows available for execution.
Available Pipelines¶
View available pipelines in the Compute service:
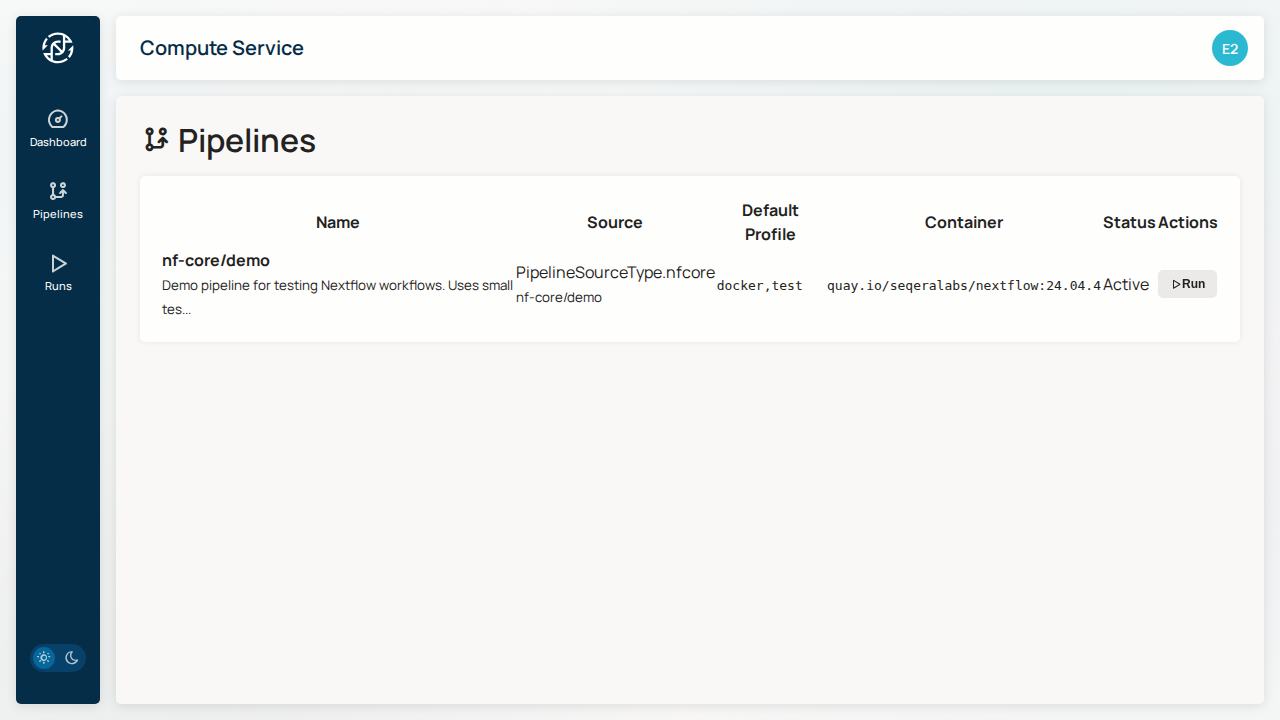
Pipeline Information¶
Each pipeline displays:
| Field | Description |
|---|---|
| Name | Pipeline identifier (e.g., nf-core/rnaseq) |
| Version | Specific version or tag |
| Description | What the pipeline does |
| Documentation | Link to pipeline docs |
nf-core Pipelines¶
Flow supports nf-core pipelines, a community collection of curated bioinformatics workflows:
- nf-core/rnaseq - RNA sequencing analysis
- nf-core/sarek - Variant calling
- nf-core/ampliseq - Amplicon sequencing
- nf-core/demo - Test pipeline for validation
Pipeline Parameters¶
Each pipeline has configurable parameters:
Required Parameters¶
Parameters that must be set before running:
- Input - Sample sheet or input files path
- Outdir - Output directory for results
Optional Parameters¶
Additional configuration options:
- Genome - Reference genome (e.g.,
GRCh38) - Profile - Execution profile (e.g.,
docker,singularity)
Running a Pipeline¶
- Click on a pipeline to view details
- Configure required parameters
- Click Launch to start execution
See Running Workflows for detailed instructions.
Custom Pipelines¶
Coming Soon
Support for custom pipelines will be added in a future release.
Next Steps¶
- Running Workflows - Execute a pipeline
- Monitoring - Track execution progress