Your First Workflow¶
This guide walks you through running your first bioinformatics pipeline on Flow.
Prerequisites¶
Before starting, ensure you have:
- Logged into the Admin Console or CLI
- Selected an organization and project
- Cloud credentials configured for your project
Using the CLI (Recommended)¶
The CLI provides the fastest way to run workflows.
Step 1: Setup¶
# Install if not already done
pip install iseq-flow
# Login and select project
flow login --token YOUR_PAT_TOKEN
flow config select-project
Step 2: View Available Pipelines¶
Output:
SLUG NAME MODE ACTIVE
--------------------------------------------------------------------------------
hereditary-mock Hereditary Mock Pipeline direct_wdl Yes
wdl-minimal WDL Minimal Pipeline direct_wdl Yes
nextflow-minimal nextflow-minimal container_entrypoint Yes
nfcore-demo nf-core/demo direct_nextflow Yes
Step 3: Get Pipeline Details¶
Output:
Pipeline: Hereditary Mock Pipeline
Slug: hereditary-mock
Type: direct_wdl
Container: broadinstitute/cromwell:86
Tags: wdl, hereditary, mock, cromwell
Description: Mock hereditary analysis pipeline for WDL/Cromwell testing.
Required inputs: case_id, child_fastq
Step 4: Submit a Run¶
File paths are relative to your project bucket:
flow runs submit --pipeline hereditary-mock \
-P case_id=test-case-001 \
-P child_fastq=data/R1.fastq.gz \
-P child_fastq=data/R2.fastq.gz \
--watch
The --watch flag monitors progress until completion:
Run submitted successfully!
ID: abc123-def456-...
Name: hereditary-mock-20260113-120000
Status: queued
Watching run status (Ctrl+C to stop)...
[12:00:15] Status: queued
[12:01:00] Status: running
Started: 2026-01-13T12:00:55Z
[12:05:00] Status: succeeded
Run completed successfully!
Output: gs://your-bucket/outputs/hereditary-mock-20260113-120000/
Step 5: Check Run Status¶
Step 6: Download Results¶
# List output files
flow files ls outputs/
# Download specific files
flow files download outputs/report.html
flow files download outputs/variants.vcf
Using the Web Interface¶
Step 1: Navigate to Compute¶
Go to the Compute service from the navigation menu.
Step 2: View Available Pipelines¶
Click on Pipelines to see available workflows:
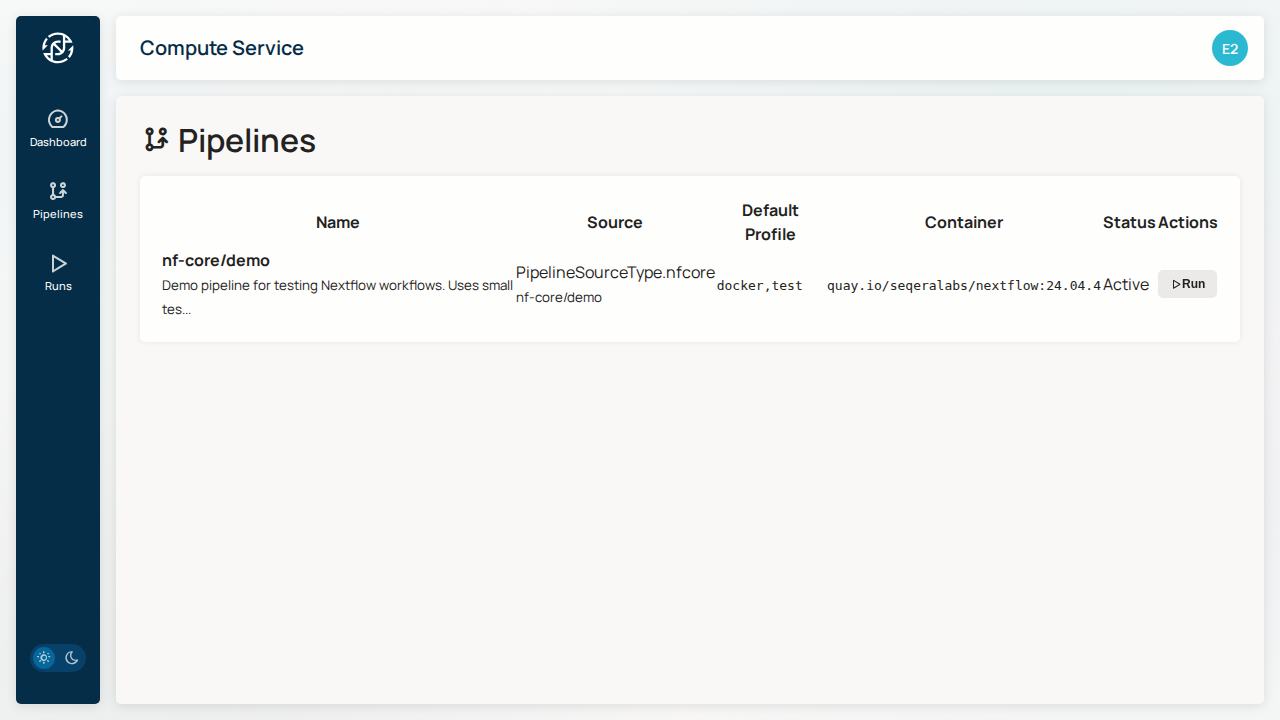
Step 3: Launch a Run¶
- Click on a pipeline to view its details
- Click Run Pipeline
- Configure run parameters:
- Name: A descriptive name for this run
- Profile: Use
docker,testfor testing -
Container: Leave as default
-
Click Submit
Step 4: Monitor Progress¶
Navigate to Runs to monitor your workflow:
Run statuses:
| Status | Description |
|---|---|
| PENDING | Run submitted, waiting to start |
| QUEUED | Queued on GCP Batch |
| RUNNING | Workflow executing |
| SUCCEEDED | Completed successfully |
| FAILED | Completed with errors |
Step 5: View Results¶
Once complete, click on the run to view:
- Execution logs
- Output file locations
- Timing and resource usage
Clinical Workflow Example¶
For clinical labs, associate runs with orders:
# 1. Create a clinical order
flow orders create -n "Patient Case 001" \
--accession "ACC-2026-001" \
--priority urgent \
--indication "Suspected hereditary cancer"
# Order created with ID: order-123...
# 2. Submit run linked to order (paths relative to bucket)
flow runs submit --pipeline hereditary-mock \
--order-id order-123... \
-P case_id=patient-001 \
-P child_fastq=data/R1.fastq.gz \
-P child_fastq=data/R2.fastq.gz \
--watch
# 3. Transition order status
flow orders transition order-123... analyzing
# 4. List runs for order
flow runs list --order-id order-123...
Next Steps¶
- Pipelines Guide - Learn about available pipelines
- Monitoring Guide - Advanced monitoring features
- File Browser - Access output files
- Managing Orders - Clinical order workflows